Gherman Novakovsky
@gnovakovsky.bsky.social
160 followers
140 following
20 posts
PhD, Illumina AI lab
Posts
Media
Videos
Starter Packs
Pinned

Reposted by Gherman Novakovsky


Reposted by Gherman Novakovsky



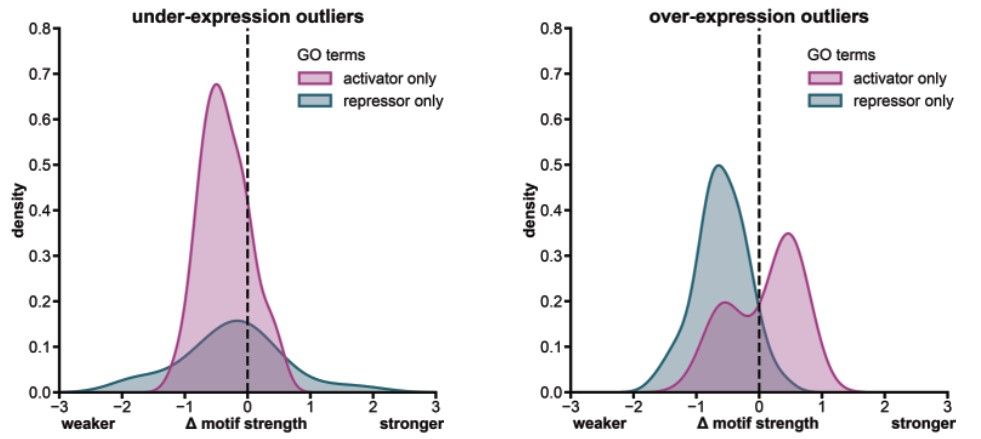
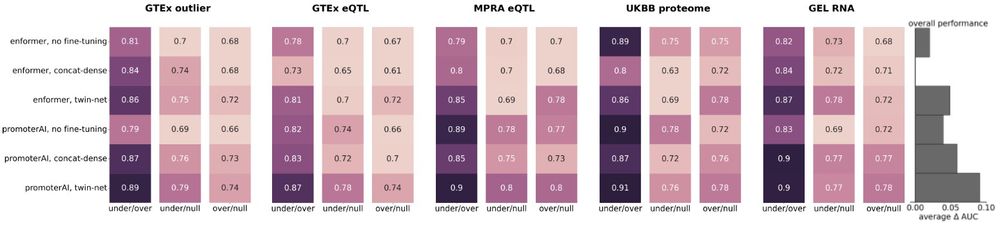
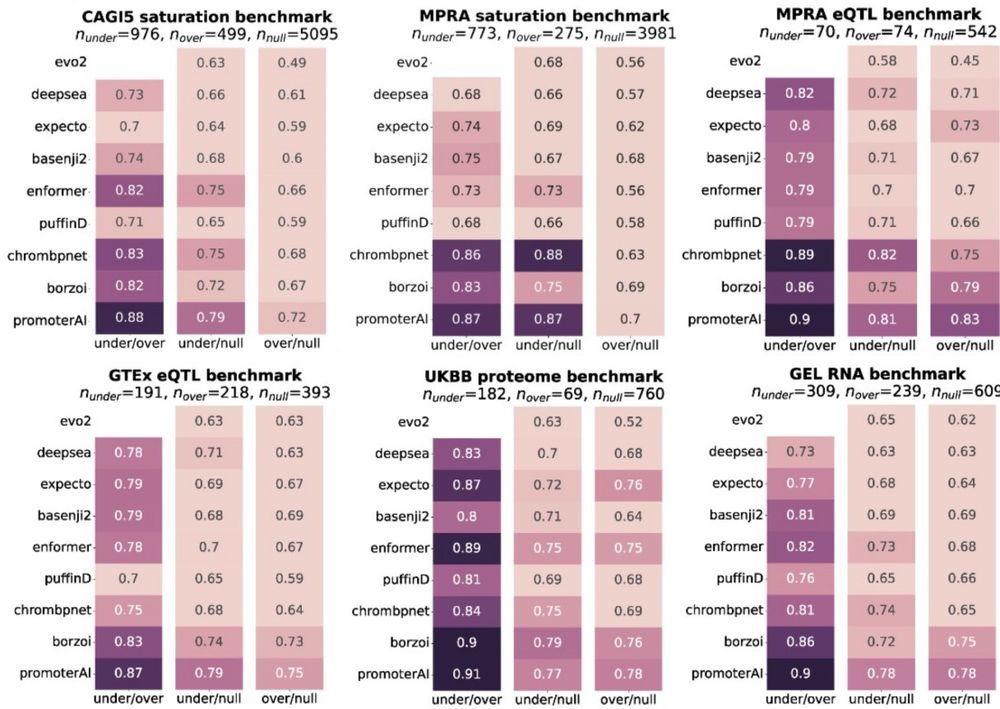
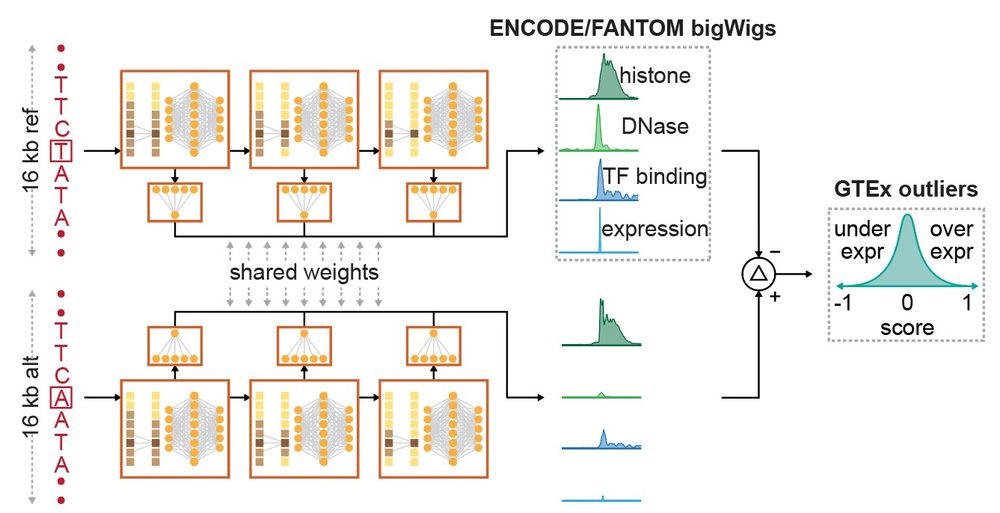
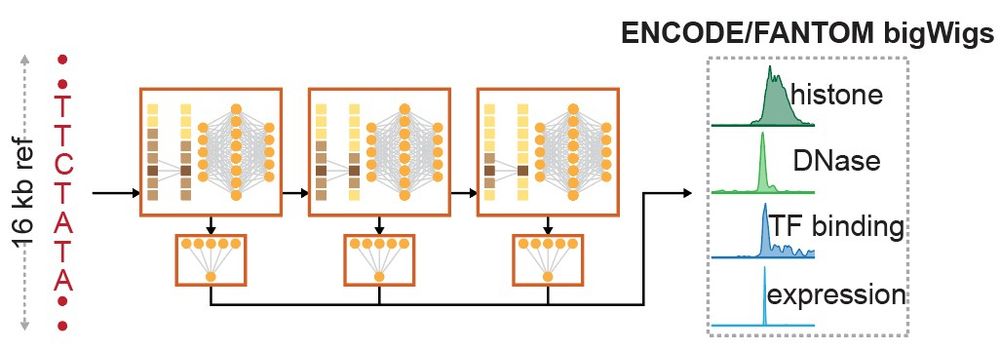
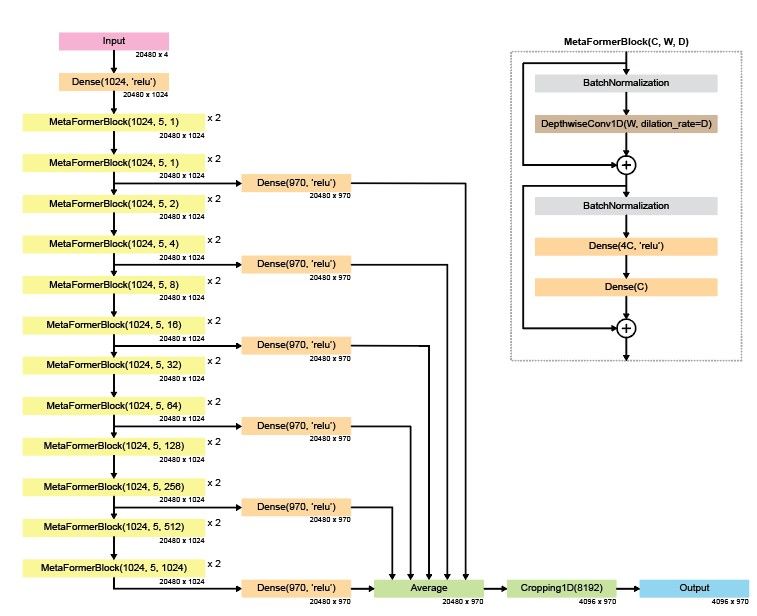
Reposted by Gherman Novakovsky
Stein Aerts
@steinaerts.bsky.social
· May 16

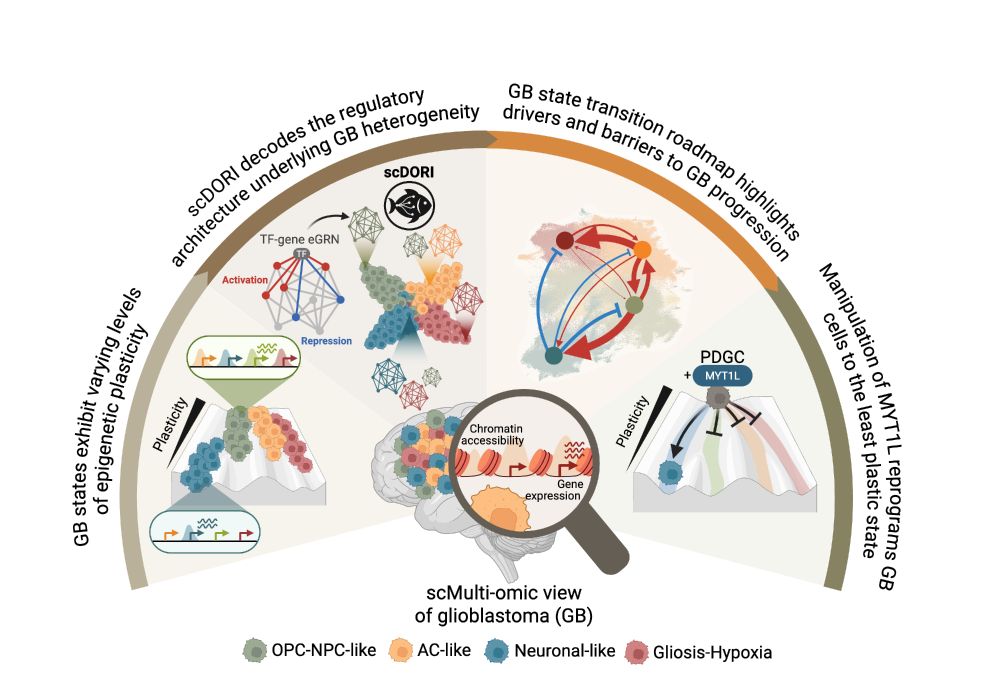
Decoding Plasticity Regulators and Transition Trajectories in Glioblastoma with Single-cell Multiomics
Glioblastoma (GB) is one of the most lethal human cancers, marked by profound intratumoral heterogeneity and near-universal treatment resistance. Cellular plasticity, the capacity of cancer cells to t...
www.biorxiv.org
Reposted by Gherman Novakovsky

Reposted by Gherman Novakovsky

Haky Im
@hakyim.bsky.social
· May 14
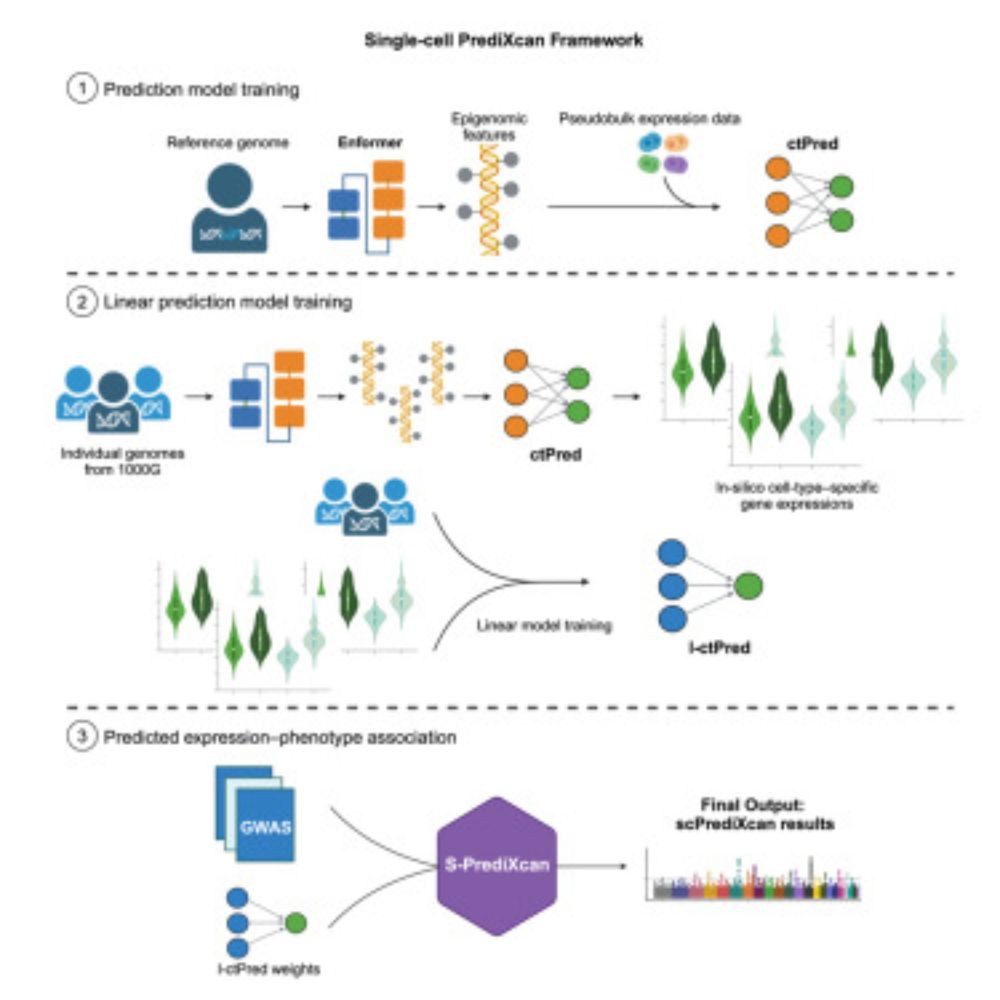
scPrediXcan integrates deep learning methods and single-cell data into a cell-type-specific transcriptome-wide association study framework
Zhou et al. introduce scPrediXcan, a novel transcriptome-wide association study framework
that integrates the deep learning-based model ctPred for cell-type-specific expression
prediction. Applied to ...
www.cell.com