Josipa Lipovac
@jlipovac.bsky.social
62 followers
87 following
13 posts
PhD student at FER, University of Zagreb | Bioinformatics | Metagenome analysis | Genome assembly
Posts
Media
Videos
Starter Packs
Pinned

Josipa Lipovac
@jlipovac.bsky.social
· May 16




Reposted by Josipa Lipovac


Reposted by Josipa Lipovac

Reposted by Josipa Lipovac

Reposted by Josipa Lipovac


Reposted by Josipa Lipovac

Josipa Lipovac
@jlipovac.bsky.social
· Jul 20

Mile Sikic
@msikic.bsky.social
· Jul 19

A Complete Telomere-to-Telomere Diploid Reference Genome for Indian Population
Human reference genomes have been instrumental in advancing genomic and biomedical research, but South and Southeast Asian populations are underrepresented, despite accounting for a large proportion o...
biorxiv.org
Reposted by Josipa Lipovac

Reposted by Josipa Lipovac


Josipa Lipovac
@jlipovac.bsky.social
· May 16
Reposted by Josipa Lipovac


Josipa Lipovac
@jlipovac.bsky.social
· May 16

Josipa Lipovac
@jlipovac.bsky.social
· May 16

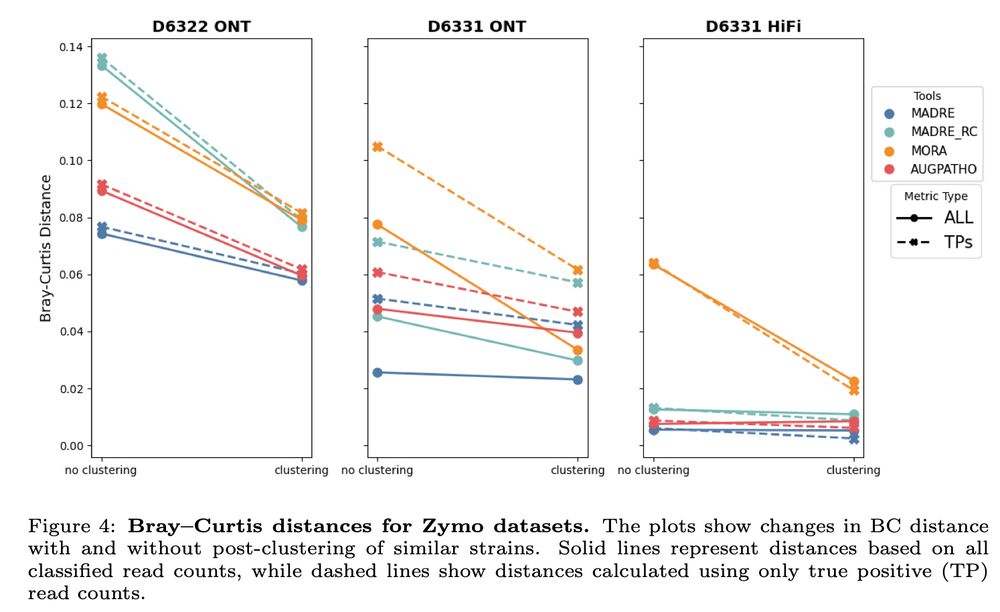


Josipa Lipovac
@jlipovac.bsky.social
· May 16
Josipa Lipovac
@jlipovac.bsky.social
· May 16
Josipa Lipovac
@jlipovac.bsky.social
· May 16
Josipa Lipovac
@jlipovac.bsky.social
· May 16
Reposted by Josipa Lipovac
Reposted by Josipa Lipovac


Reposted by Josipa Lipovac
Zamin Iqbal
@zaminiqbal.bsky.social
· Feb 28

Are reads required? High-precision variant calling from bacterial genome assemblies
Accurate nucleotide variant calling is essential in microbial genomics, particularly for outbreak tracking and phylogenetics. This study evaluates variant calls derived from genome assemblies compared...
www.biorxiv.org