Roland Faure
@rfaure.bsky.social
54 followers
130 following
11 posts
Sequence bioinfomatician, algorithms, methods.
Postdoc in Institut Pasteur in Rayan Chikhi's lab
Posts
Media
Videos
Starter Packs




Reposted by Roland Faure


Reposted by Roland Faure


Roland Faure
@rfaure.bsky.social
· May 16
Reposted by Roland Faure

Josipa Lipovac
@jlipovac.bsky.social
· May 16
Reposted by Roland Faure


Reposted by Roland Faure


Reposted by Roland Faure

Zamin Iqbal
@zaminiqbal.bsky.social
· Apr 9
Michael Baym
@baym.lol
· Apr 9
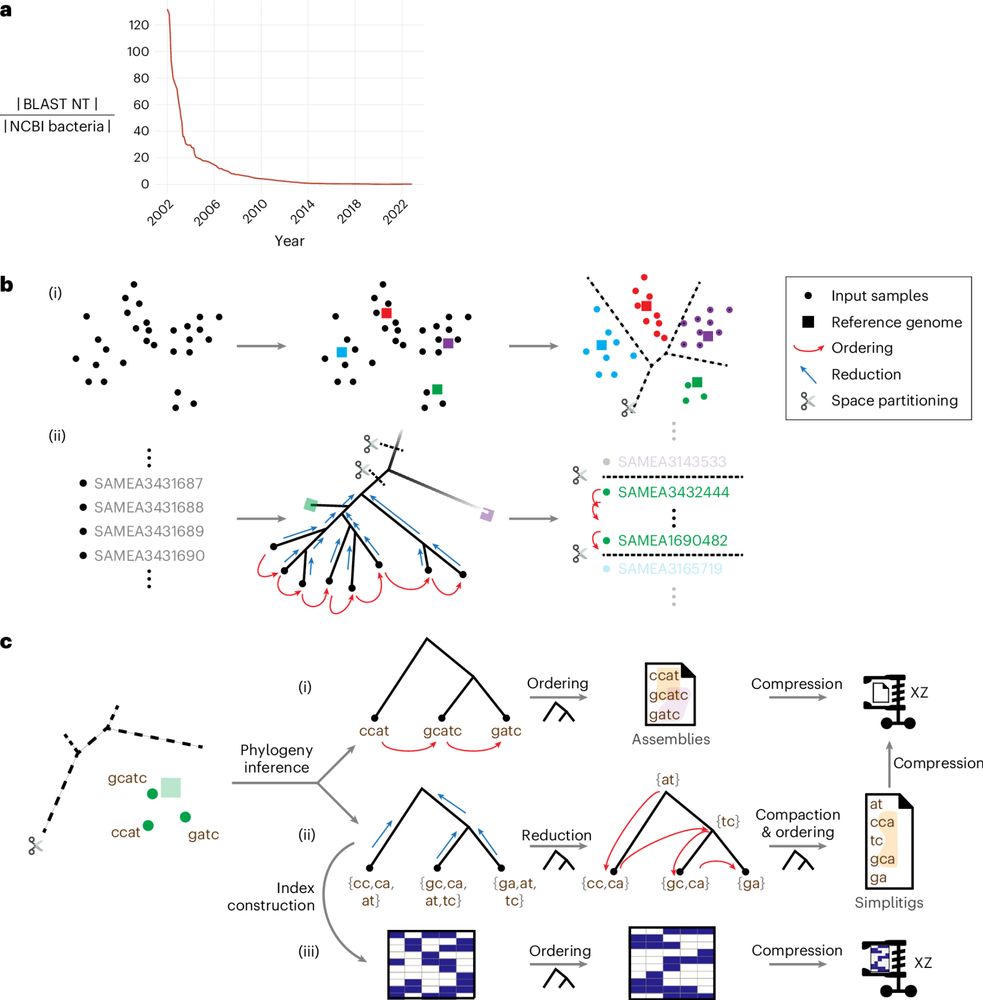
Efficient and robust search of microbial genomes via phylogenetic compression
Nature Methods - Phylogenetic compression achieves performant and lossless compression of massive collections of microbial genomes, facilitating fast BLAST-like search and versatile alignment tasks.
rdcu.be


Reposted by Roland Faure

Igor Martayan
@imartayan.bsky.social
· Dec 13

Ragnar {Groot Koerkamp}
@curiouscoding.nl
· Dec 13
Roland Faure
@rfaure.bsky.social
· Dec 13
Reposted by Roland Faure



Roland Faure
@rfaure.bsky.social
· Dec 2