Leandros Boukas
@leandrosboukas.bsky.social
140 followers
250 following
28 posts
Epigenetics/Gene regulation, Human Genetics, Population Genetics. Medical Genetics & Genomics Fellow at Harvard Medical School and Boston Children's Hospital. MD, PhD
Posts
Media
Videos
Starter Packs


Reposted by Leandros Boukas

John Greally
@greally.bsky.social
· Aug 2
Reposted by Leandros Boukas



Reposted by Leandros Boukas

Reposted by Leandros Boukas

Richard Sever
@richardsever.bsky.social
· May 18






Reposted by Leandros Boukas

Lars Velten
@larsplus.bsky.social
· May 8
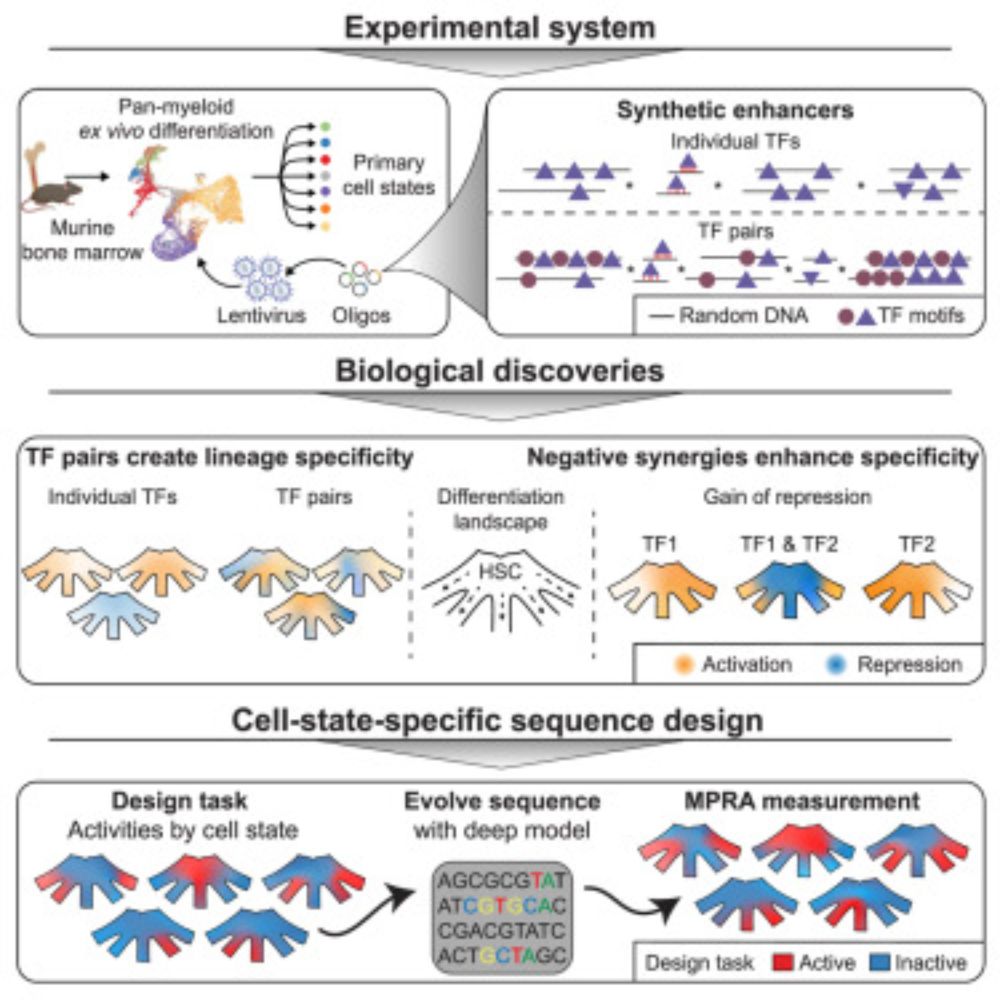
Design principles of cell-state-specific enhancers in hematopoiesis
Screen of minimalistic enhancers in blood progenitor cells demonstrates widespread
dual activator-repressor function of transcription factors (TFs) and enables the model-guided
design of cell-state-sp...
www.cell.com
Reposted by Leandros Boukas

Reposted by Leandros Boukas

Michael Love
@mikelove.bsky.social
· Apr 8
Reposted by Leandros Boukas

Reposted by Leandros Boukas

Gang Fang
@gangfang.bsky.social
· Mar 30





Reposted by Leandros Boukas

Tuuli Lappalainen
@tuuliel.bsky.social
· Mar 19
Reposted by Leandros Boukas

Nikolai Slavov
@slavov-n.bsky.social
· Mar 1

Protein degradation and growth dependent dilution substantially shape mammalian proteomes
Cellular protein concentrations are maintained through a balance of synthesis and clearance. Clearance occurs through both protein degradation and growth-dependent dilution. At slow growth, clearance ...
www.biorxiv.org
Reposted by Leandros Boukas

Wolfgang Huber
@wkhuber.bsky.social
· Feb 27