Vikram Shivakumar
@vikramshivakumar.bsky.social
130 followers
110 following
19 posts
PhD Student @ JHU Langmead Lab
Posts
Media
Videos
Starter Packs
Reposted by Vikram Shivakumar


Reposted by Vikram Shivakumar




Reposted by Vikram Shivakumar


Reposted by Vikram Shivakumar

Liana Lareau
@lianafaye.bsky.social
· Aug 7

Protein language models reveal evolutionary constraints on synonymous codon choice
Evolution has shaped the genetic code, with subtle pressures leading to preferences for some synonymous codons over others. Codons are translated at different speeds by the ribosome, imposing constrai...
www.biorxiv.org
Reposted by Vikram Shivakumar


Reposted by Vikram Shivakumar


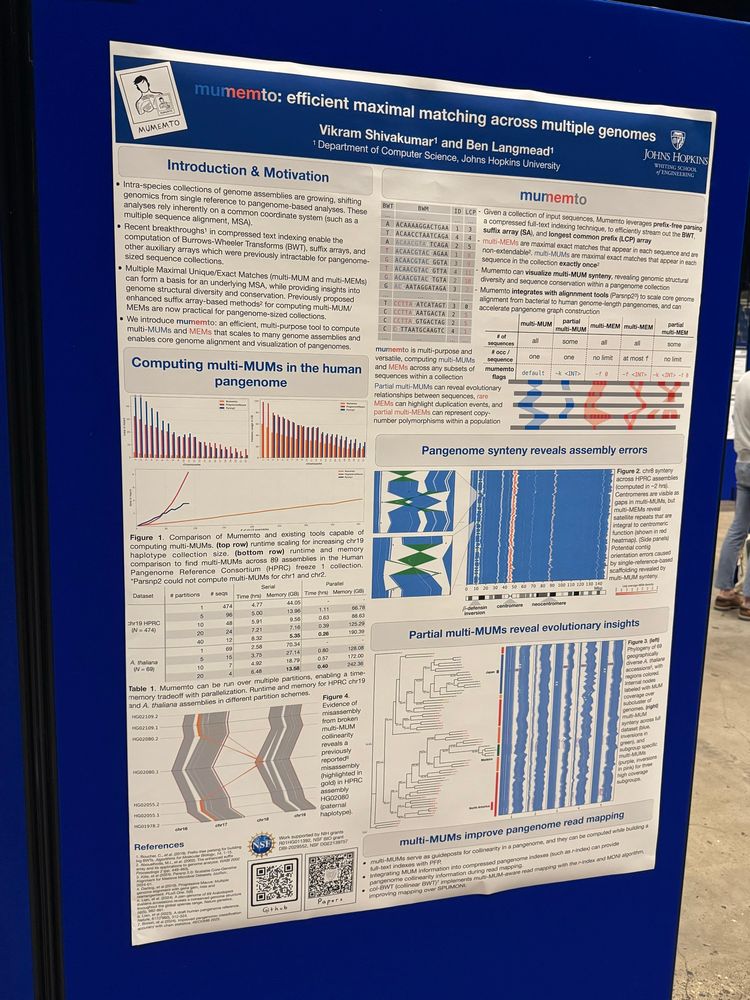

Ben Langmead
@benlangmead.bsky.social
· Jun 17
Reposted by Vikram Shivakumar
Reposted by Vikram Shivakumar

Mohsen Zakeri
@mohsenzakeri.bsky.social
· May 29
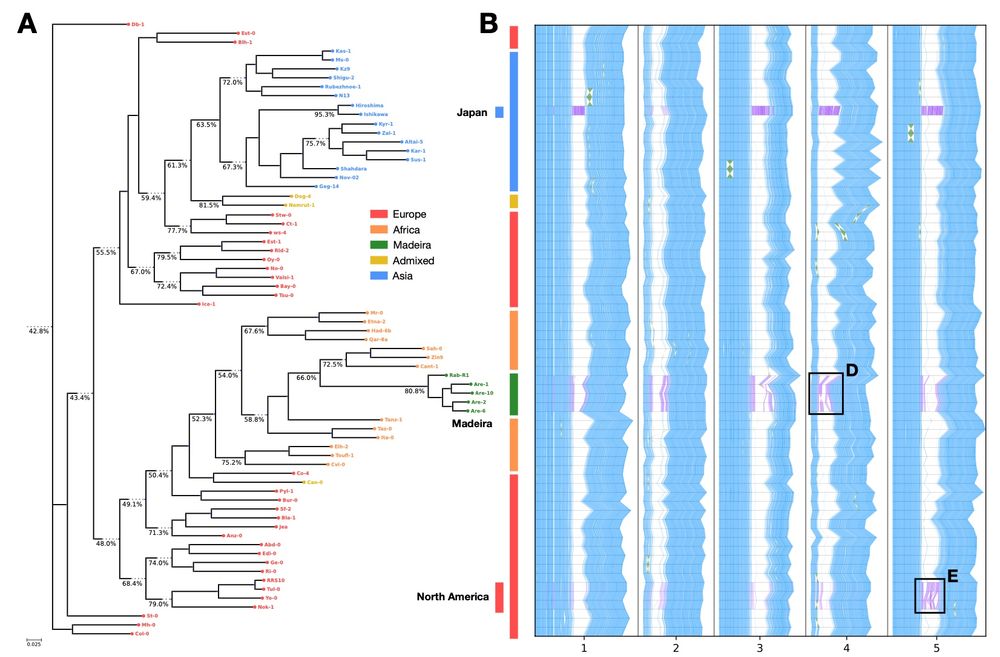
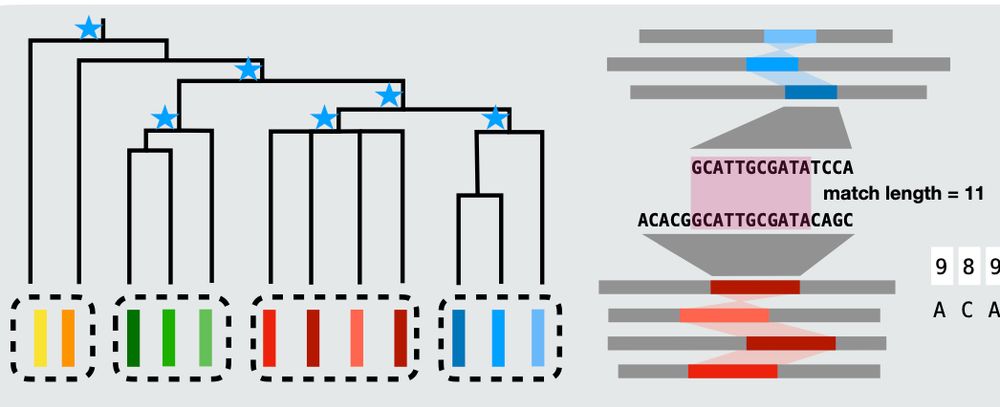

Reposted by Vikram Shivakumar

Arun Das
@arun-das.bsky.social
· May 15

Reposted by Vikram Shivakumar

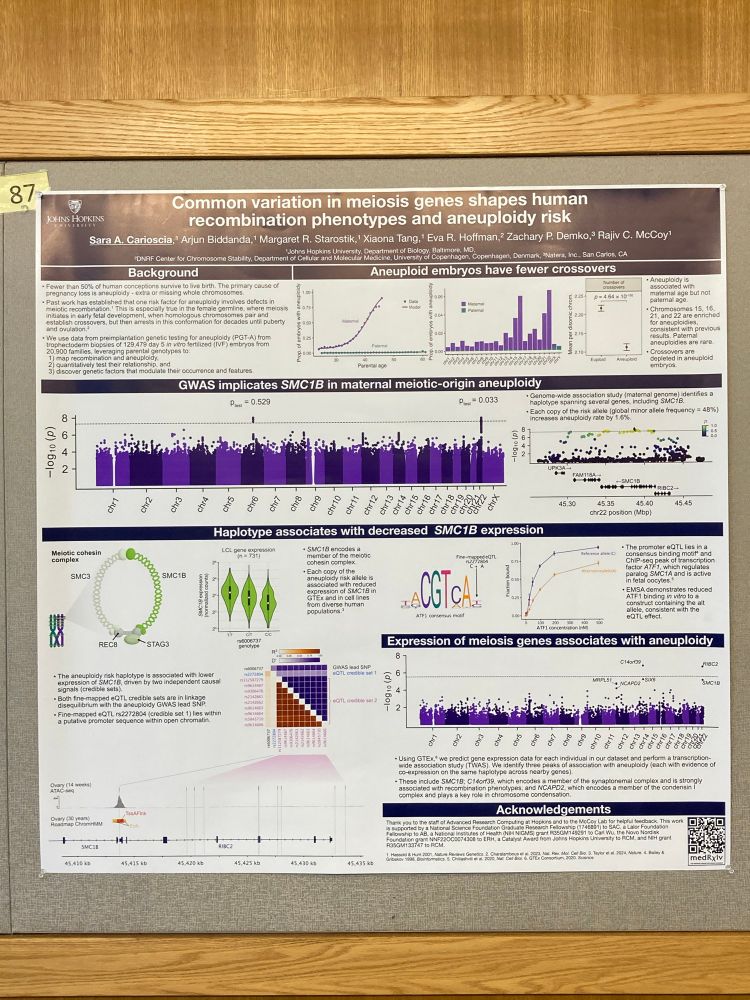
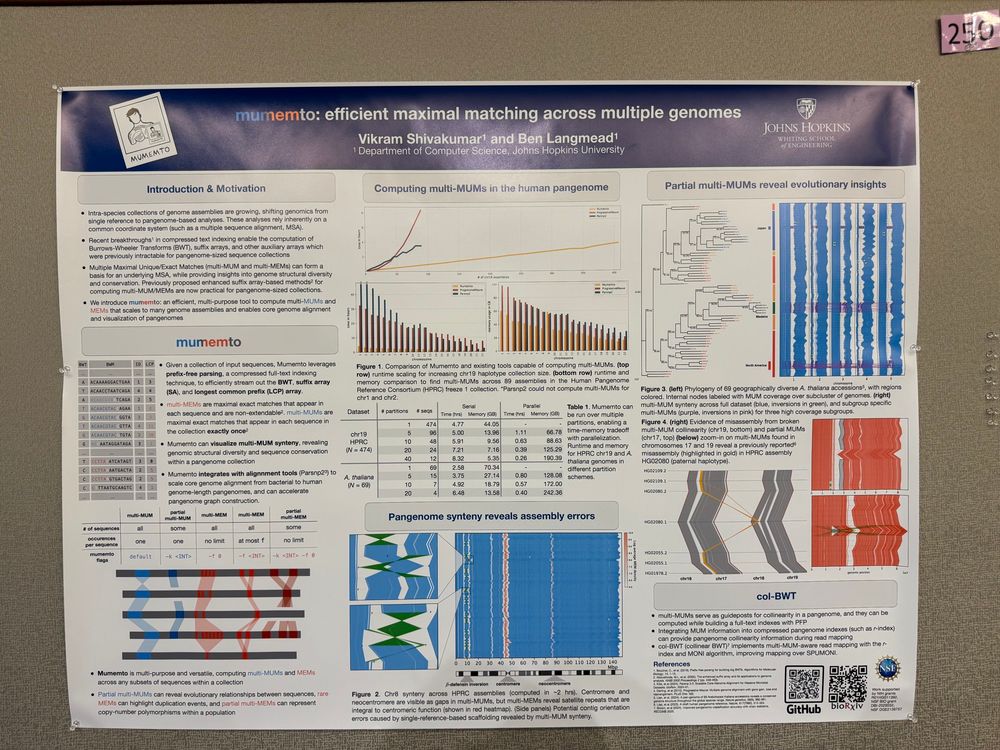
Reposted by Vikram Shivakumar


Reposted by Vikram Shivakumar

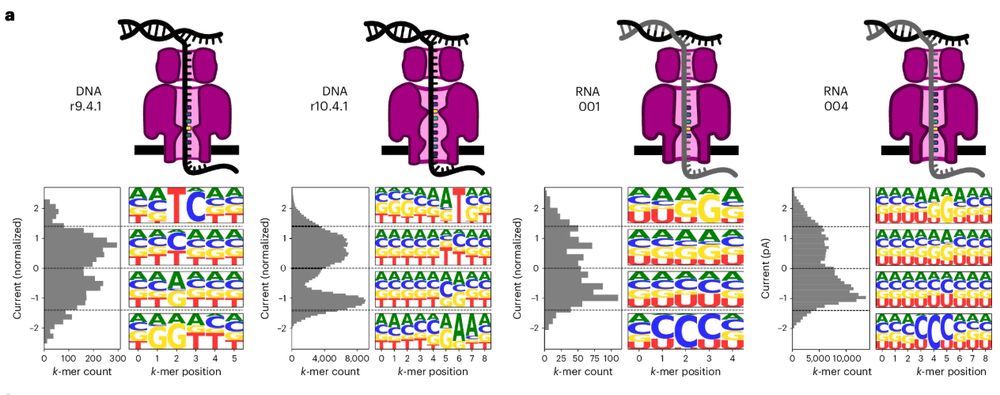
Reposted by Vikram Shivakumar

Bohan Ni
@bohanni.bsky.social
· Mar 26
Integration of transcriptomics and long-read genomics prioritizes structural variants in rare disease
An international, peer-reviewed genome sciences journal featuring outstanding original research that offers novel insights into the biology of all organisms
genome.cshlp.org